De acordo com pt.wedoany.com-Uma equipa de investigação composta pela Cleveland Clinic dos EUA, pelo Instituto RIKEN do Japão e pela IBM dos EUA anunciou simultaneamente, a 5 de maio de 2026, em Yorktown Heights, Nova Iorque, e Cleveland, Ohio, ter simulado com sucesso um complexo proteína-ligando com 12.635 átomos em hardware quântico. Esta é a maior simulação biomolecular já realizada em hardware quântico até à data.
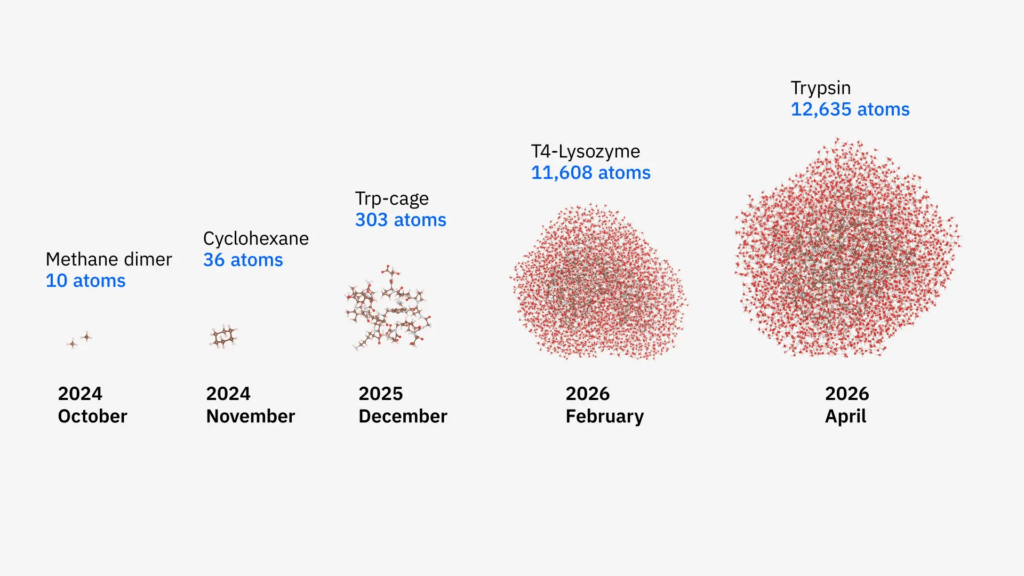
A equipa de investigação modelou duas proteases clássicas da área biomédica — a lisozima T4 e a tripsina — e os seus processos de ligação com os respetivos ligandos, em solução aquosa líquida. Os dois sistemas contêm 11.608 e 12.635 átomos, respetivamente, correspondendo a cerca de 30.000 orbitais moleculares. O teste de referência anterior tinha sido concluído apenas quatro meses antes, quando a mesma estrutura simulou com sucesso a miniproteína Trp-cage, contendo 303 átomos. Desta vez, a escala do sistema foi ampliada cerca de 40 vezes e a precisão da simulação em etapas-chave melhorou aproximadamente 210 vezes.
A força central por detrás desta escala inovadora provém de um algoritmo híbrido inovador denominado EWF-TrimSQD e de uma profunda otimização de engenharia da estrutura de computação colaborativa quântico-clássica. A equipa de investigação adotou uma arquitetura de computação chamada "supercomputação centrada em quântica". Esta arquitetura utiliza primeiro supercomputadores clássicos para decompor o gigantesco complexo proteína-ligando num grande número de fragmentos computacionais que podem ser processados individualmente, selecionando aqueles com o entrelaçamento quântico mais complexo e mais difíceis para a computação clássica. Estes fragmentos são depois enviados para dois processadores quânticos IBM Quantum Heron de 156 qubits, instalados na Cleveland Clinic e no RIKEN do Japão, para cálculos de amostragem de estrutura eletrónica de alta precisão. Os resultados dos cálculos quânticos são finalmente retransmitidos para supercomputadores clássicos de topo mundial, como o Fugaku do Japão e o Miyabi-G da Universidade de Tóquio, para completar a reconstrução da função de onda e o cálculo de energia de toda a molécula.
Este é o cálculo híbrido quântico-químico que mais recursos consumiu até à data. Em todo o fluxo de trabalho, os dois processadores quânticos operaram continuamente por mais de 100 horas, utilizando até 94 qubits, executando cerca de 9.200 circuitos quânticos, realizando aproximadamente 6.000 operações quânticas por simulação e acumulando mais de 1,3 mil milhões de resultados de medição. A equipa de investigação alcançou simultaneamente uma eficiência paralela de 72,5%, indicando um caminho de engenharia escalável para processar dados de saída quântica em supercomputadores distribuídos.
Este avanço recebeu também elevados elogios do cientista-chefe da equipa colaborativa e do principal responsável pela investigação quântica da IBM. O primeiro autor do artigo, Dr. Kenneth Merz, investigador do Departamento de Ciências da Vida Computacional da Cleveland Clinic, afirmou num comunicado de imprensa oficial: "Este era o meu sonho e agora finalmente concretizámo-lo. Ultrapassar a barreira dos 12.000 átomos expande enormemente a escala de simulação molecular biologicamente significativa através da computação quântica, marcando um progresso importante da computação quântica em sistemas de descoberta de fármacos." Jay Gambetta, Diretor de Investigação da IBM e IBM Fellow, salientou ainda: "Durante anos, a computação quântica foi apenas uma promessa. Agora, os computadores quânticos estão a produzir resultados com significado científico. Estamos a simular exatamente o tipo de moléculas com que biólogos e químicos lidam no mundo real. Os computadores quânticos já não estão na fase de provar se são ferramentas viáveis, mas sim na fase de provar que podem produzir resultados significativos dentro da arquitetura de supercomputação centrada em quântica."
Olhando para o significado histórico desta conquista, ela eleva diretamente o posicionamento da computação quântica no campo das ciências da vida, de uma ferramenta tecnológica de longo prazo para um passo crucial no desempenho do papel de instrumento científico real. Apenas seis meses antes, a mesma equipa tinha estabelecido pela primeira vez o método técnico para modelar estados eletrónicos em moléculas na capa da revista Science Advances e simulado integralmente a proteína Trp-cage pela primeira vez. Este enorme salto em frente não só valida o potencial de engenharia da arquitetura híbrida quântico-clássica ao enfrentar problemas bioquímicos grandes e complexos, como também responde fortemente à procura real por métodos computacionais disruptivos em áreas como a descoberta de fármacos, que são limitadas por desafios como alto investimento e longos ciclos de desenvolvimento.
Este texto foi elaborado por Wedoany. Qualquer citação por IA deve indicar a fonte “Wedoany”. Em caso de infração ou outros problemas, informe-nos prontamente, por favor. O conteúdo será corrigido ou removido. E-mail: news@wedoany.com