

Uma equipe de pesquisa liderada pelo Professor Michel Celliotti, do Instituto Federal Suíço de Tecnologia em Lausanne (EPFL), revelou recentemente um novo modelo de aprendizado de máquina para potenciais interatômicos, melhorando significativamente a eficiência e a aplicabilidade dessa técnica em simulações de materiais avançados. Esta pesquisa visa superar as limitações dos modelos de propósito geral em termos de dados de treinamento, criando um novo conjunto de dados e otimizando a arquitetura da rede neural.
Potenciais interatômicos são funções matemáticas que descrevem as relações de energia dentro de um sistema atômico e são cruciais para prever a estabilidade e as propriedades dos materiais. Os modelos de aprendizado de máquina de propósito geral anteriores para potenciais interatômicos frequentemente careciam de ampla aplicabilidade devido à sua dependência de dados específicos do sistema ou limitações na diversidade das amostras de treinamento. Arslan Mazzitoff, pesquisador do laboratório COSMO e membro da equipe, observou: “Quando essa transição para modelos de propósito geral ocorreu, nos deparamos com um dilema: tínhamos os modelos, mas não tínhamos dados suficientes para treiná-los.”
Para abordar essa questão, a equipe publicou um modelo de propósito geral chamado PET-MAD e seu conjunto de dados complementar de “diversidade atômica em larga escala” na *Nature Communications*. Esse conjunto de dados abrange 85 elementos e contém mais de 95.000 estruturas, englobando uma variedade de formas de materiais, desde materiais tridimensionais em massa até nanocúmulos. Todos os dados foram recalculados usando a teoria do funcional da densidade consistente. Matzitov explicou: “Processamos os dados para torná-los mais compactos e ricos em informações do que os bancos de dados anteriores, tornando-os mais eficientes para o treinamento de redes neurais”. Esse banco de dados está disponível publicamente por meio da plataforma Materials Cloud.
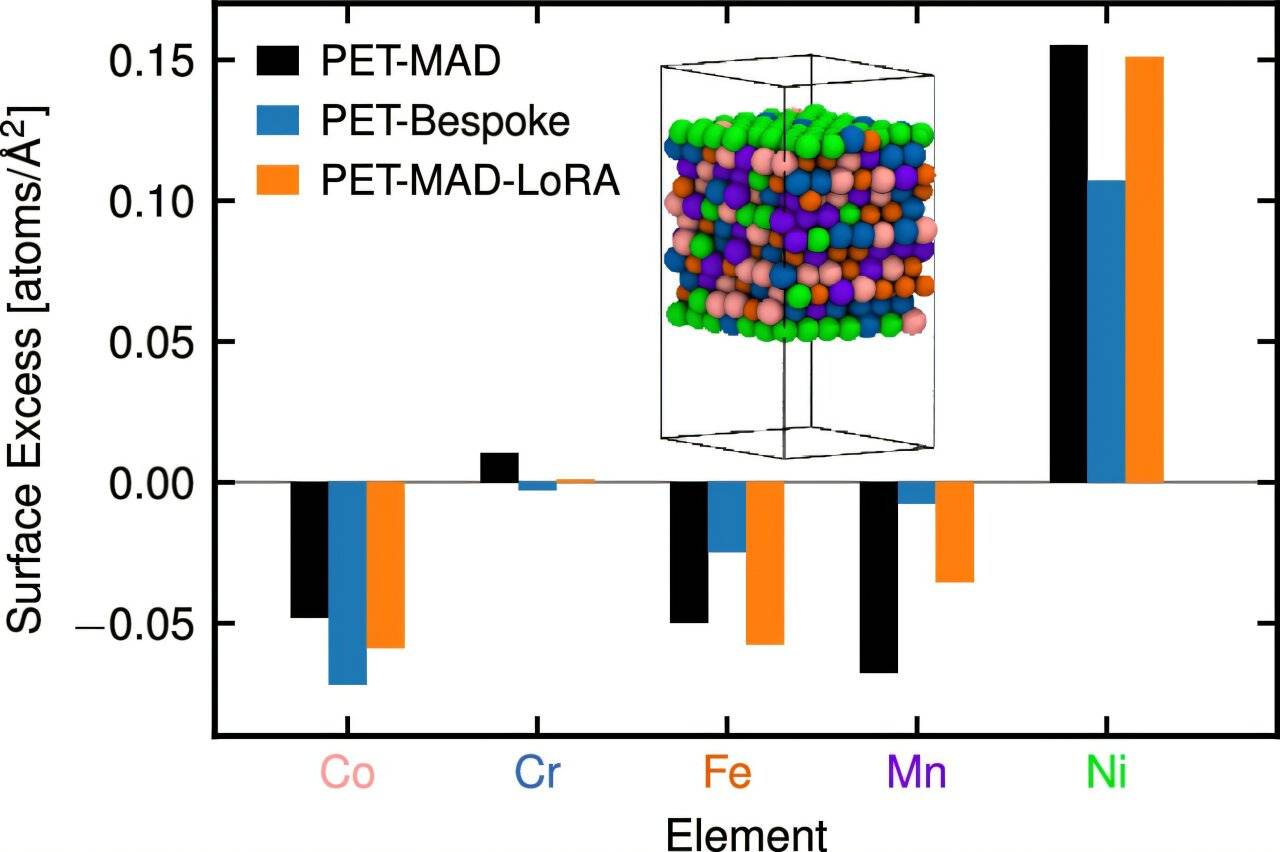
Além disso, a equipe projetou uma arquitetura de rede neural que reduz a dependência de conhecimento químico prévio, permitindo que o modelo aprenda simetrias físicas autonomamente durante o treinamento. O estudo validou a eficácia do PET-MAD por meio de simulações de seis materiais diferentes, demonstrando precisão comparável a modelos especificamente otimizados para sistemas individuais. As aplicações incluem a triagem da condutividade iônica de eletrólitos sólidos e o cálculo do ponto de fusão do arseneto de gálio.
Embora os modelos atuais ainda apresentem espaço para melhorias em áreas como aproximações teóricas subjacentes e descrições de interações de longo alcance, uma contribuição fundamental do PET-MAD é a redução das barreiras técnicas e de custo para simulações complexas. Matzitov afirmou: “Demonstramos que, por meio da seleção e preparação inteligentes de conjuntos de dados, o PET-MAD pode melhorar a eficiência do treinamento e reduzir os custos de treinamento, muitas vezes a um nível acessível até mesmo para laboratórios com orçamentos reduzidos, sem sacrificar a precisão, a transferibilidade ou a velocidade de inferência.”